DEBUG:coordination_geometry_finder:compute_structure_environments:Getting DetailedVoronoiContainer
DEBUG:voronoi:__init__:Setting Voronoi list
DEBUG:voronoi:setup_voronoi_list:Getting all neighbors in structure
DEBUG:voronoi:setup_voronoi_list:Setting up Voronoi list :
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #0 (1/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #1 (2/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #2 (3/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #3 (4/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #4 (5/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #5 (6/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #6 (7/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #7 (8/9)
DEBUG:voronoi:setup_voronoi_list: - Voronoi analysis for site #8 (9/9)
DEBUG:voronoi:setup_voronoi_list:Voronoi list set up in 0.27 seconds
DEBUG:voronoi:__init__:Setting neighbors distances and angles
DEBUG:voronoi:__init__:Neighbors distances and angles set up in 0.00 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments:DetailedVoronoiContainer has been set up
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #0/9 (Si)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (4, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.03 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #1/9 (Si)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (4, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.03 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #2/9 (Si)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (4, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.03 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #3/9 (O)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (2, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.02 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #4/9 (O)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (2, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.02 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #5/9 (O)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (2, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.02 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #6/9 (O)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (2, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.02 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #7/9 (O)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (2, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.02 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... in site #8/9 (O)
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_set (2, 0)
DEBUG:coordination_geometry_finder:update_nb_set_environments:Getting StructureEnvironments with optimized algorithm
DEBUG:coordination_geometry_finder:compute_structure_environments: ... getting environments for nb_sets added from hints
DEBUG:coordination_geometry_finder:compute_structure_environments: ... computed in 0.02 seconds
DEBUG:coordination_geometry_finder:compute_structure_environments: ... compute_structure_environments ended in 0.52 seconds
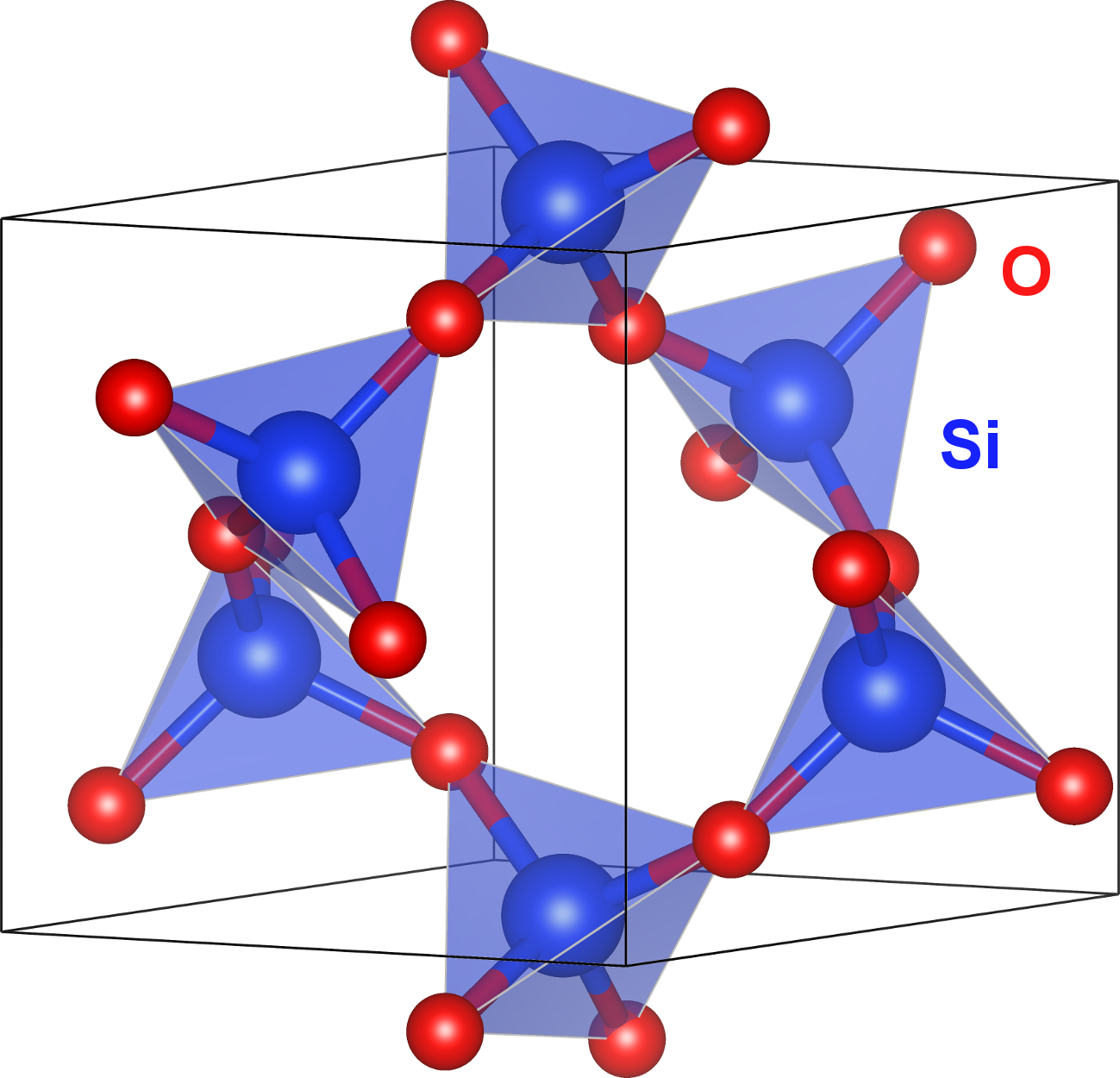 The graphic is created with VESTA: K. Momma and F. Izumi, "VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data," J. Appl. Cryst., 2011, 44, 1272.
The graphic is created with VESTA: K. Momma and F. Izumi, "VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data," J. Appl. Cryst., 2011, 44, 1272.